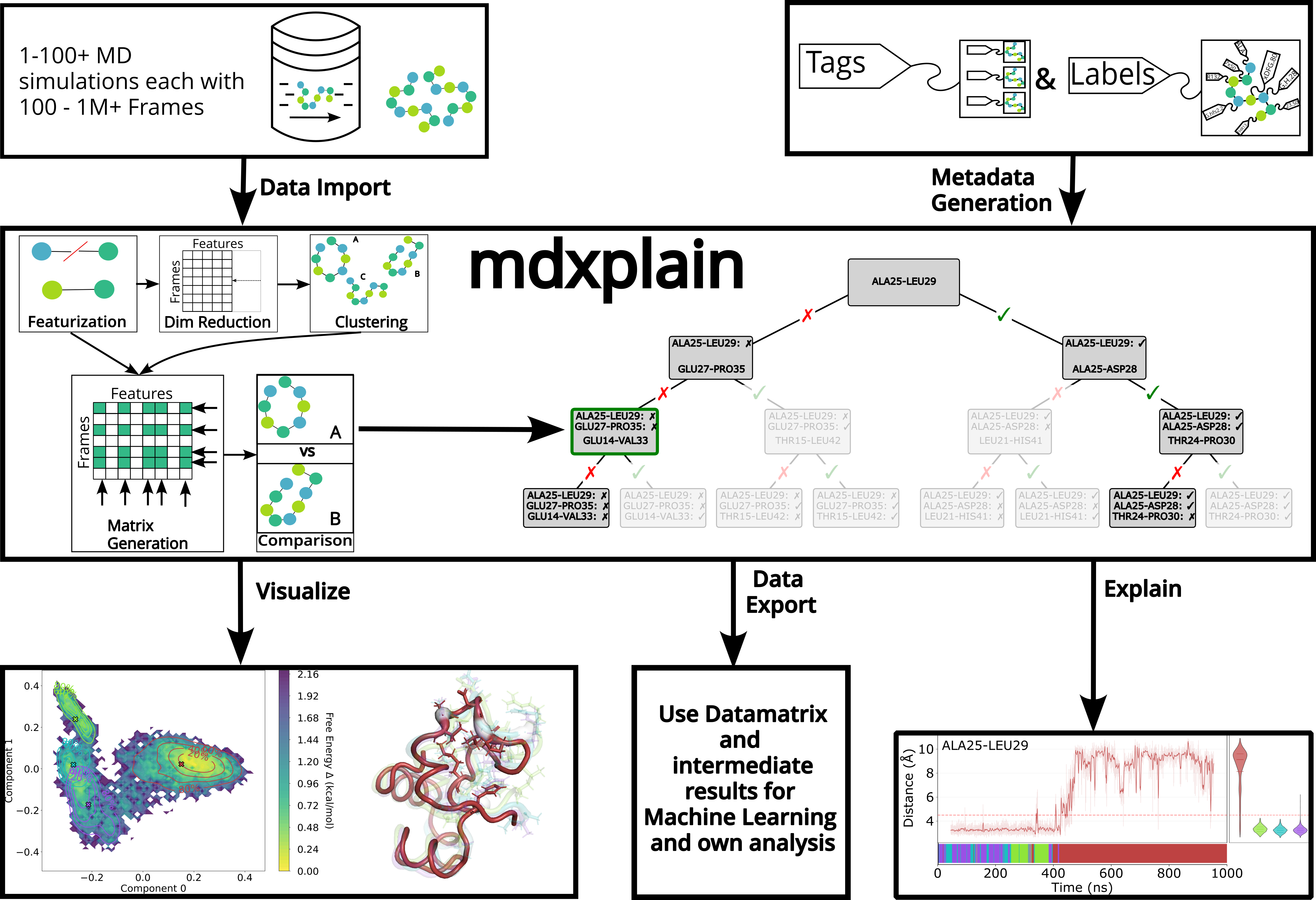
Welcome to mdxplain
A Python toolkit designed for interpretable molecular dynamics trajectory analysis, efficiently processing large datasets while delivering understandable results through machine learning models. It combines modular workflows with memory-efficient processing and decision trees to identify key conformational features and streamline complex analytical pipelines.
You can check and download the code on GitHub mdxplain.
Quick Start Installation
mdxplain uses a Makefile for streamlined installation and development workflows. For detailed instructions, please refer to the How to Install mdxplain guide.
git clone https://github.com/maximilian-salomon/mdxplain.git
cd mdxplain
Conda Environment (Recommended)
Create a new conda environment with production dependencies, Jupyter, and nglview:
make setup-conda
conda activate mdxplain
Python Virtual Environment (Alternative)
Create a new virtual environment with production dependencies, Jupyter, and nglview:
make setup-venv
source mdxplain-venv/bin/activate
Install in Existing Environment
Install mdxplain core package in your currently active environment:
# Without Jupyter notebooks and nglview
make install
# With Jupyter notebooks and nglview
make install-jupyter
Optional: PyMOL Installation
PyMOL is not included in the standard installation to avoid compatibility issues due to its complex system-level dependencies. If you need PyMOL for 3D structure visualization, install it separately:
make install-pymol
Note: For system-specific installation instructions, please refer to the official PyMOL documentation. Alternatively, you can install PyMOL independently and load mdxplain’s generated PyMOL scripts manually.
Workflow Example
This example demonstrates a complete analysis pipeline from trajectory loading to interpretable results.
Setup and Data Loading
from mdxplain import PipelineManager
# Initialize pipeline and load trajectory data
pipeline = PipelineManager(show_progress=False)
pipeline.trajectory.load_trajectories(data_input="data/2RJY")
pipeline.trajectory.add_labels(traj_selection="all")
Feature Extraction and Selection
Extract molecular features and create a custom feature subset.
# Add distance and contact features
pipeline.feature.add.distances()
pipeline.feature.add.contacts(cutoff=4.5)
# Create and configure feature selector
pipeline.feature_selector.create("contacts_only")
pipeline.feature_selector.add.contacts("contacts_only", "all")
pipeline.feature_selector.select("contacts_only")
Dimensionality Reduction and Clustering
Reduce feature space and identify conformational states.
# Apply Contact Kernel PCA
pipeline.decomposition.add.contact_kernel_pca(
n_components=4,
selection_name="contacts_only",
decomposition_name="ContactKernelPCA",
)
# Cluster using Density Peak Algorithm
pipeline.clustering.add.dpa(
"ContactKernelPCA",
Z=2.5,
cluster_name="DPA_ContactKPC"
)
Feature Importance Analysis
Identify which molecular features distinguish conformational states.
# Setup comparison groups from clusters
pipeline.data_selector.create_from_clusters(
group_name="cluster",
clustering_name="DPA_ContactKPC"
)
# Create one-vs-rest comparison
pipeline.comparison.create_comparison(
name="cluster_comparison",
mode="one_vs_rest",
feature_selector="contacts_only",
data_selector_groups="cluster"
)
# Add decision tree for interpretability
pipeline.feature_importance.add.decision_tree(
comparison_name="cluster_comparison",
analysis_name="feature_importance"
)
Visualization
# Cluster membership visualization
fig = pipeline.plots.clustering.membership(clustering_name="DPA_ContactKPC")
# Free energy landscape
fig = pipeline.plots.landscape(
decomposition_name="ContactKernelPCA",
dimensions=[0, 1]
)
# Decision tree visualization
fig = pipeline.plots.feature_importance.decision_trees(
feature_importance_name="feature_importance",
short_layout=True
)
# Feature distribution comparison
fig = pipeline.plots.feature_importance.violins(
feature_importance_name="feature_importance"
)
# Time-resolved feature importance
fig = pipeline.plots.feature_importance.time_series(
feature_importance_name="feature_importance",
membership_per_feature=True,
clustering_name="DPA_ContactKPC"
)
3D Visualization with PyMOL or NGLView
pipeline.structure_visualization.feature_importance.create_pdb_with_beta_factors(
structure_viz_name="structure_viz",
feature_importance_name="feature_importance",
)
# Create PyMOL script and call pymol structure_viz.pml
pipeline.structure_visualization.feature_importance.create_pymol_script(
structure_viz_name="structure_viz"
)
# Or use nglview in Jupyter notebooks
ui, view = pipeline.structure_visualization.feature_importance.visualize_nglview_jupyter(
structure_viz_name="structure_viz",
)